นิยามของโรคธาลัสซีเมีย
ธาลัสซีเมียเป็นโรคที่ถ่ายทอดทางพันธุกรรมซึ่งร่างกายสังเคราะห์ฮีโมโกลบินในรูปแบบที่ผิดปกติ
อย่างที่คนส่วนใหญ่ทราบ เฮโมโกลบินเป็นโปรตีนที่มีอยู่ในเซลล์เม็ดเลือดแดง ซึ่งจำเป็นสำหรับการขนส่งออกซิเจนในเลือด ในอาสาสมัครที่เป็นโรคธาลัสซีเมีย รูปแบบที่กลายพันธุ์ของเฮโมโกลบินทำให้เกิดการทำลายเซลล์เม็ดเลือดแดงอย่างค่อยเป็นค่อยไปแต่ไม่อาจหยุดยั้งได้ จนถึงภาวะโลหิตจาง
จากสถิติทางการแพทย์เห็นได้ชัดว่าโรคธาลัสซีเมียส่วนใหญ่ส่งผลกระทบต่อผู้อยู่อาศัยในประเทศแถบตะวันออกกลาง ประเทศในแอฟริกา และบรรดาผู้ที่อาศัยอยู่ตามหนองน้ำ โรคโลหิตจางเมดิเตอร์เรเนียน).
การจำแนกประเภทและสาเหตุ
ตามหน่วยย่อยโปรตีนที่บกพร่อง (ซึ่งประกอบเป็นเฮโมโกลบิน) ธาลัสซีเมียสองรูปแบบมีความโดดเด่น ก่อนดำเนินการวิเคราะห์ เราจะย้อนกลับไปชี้แจงแนวคิดที่สำคัญบางอย่าง
เฮโมโกลบินเป็นพาหะนำโรคที่ดีเลิศซึ่งใช้ในการขนส่งออกซิเจนในเลือด ประกอบด้วยโปรตีน 2 ชนิดเรียกว่าอัลฟาโกลบูลินและเบตาโกลบูลิน
ธาลัสซีเมียเกิดขึ้นเมื่อยีนหนึ่งตัวหรือมากกว่าที่ควบคุมการผลิตโปรตีนหนึ่งหรือทั้งสองชนิดมีข้อบกพร่อง (กลายพันธุ์)
ธาลัสซีเมียเกิดจากการกลายพันธุ์ของโปรตีนที่สร้างฮีโมโกลบินใน DNA: การเปลี่ยนแปลงเหล่านี้ส่งผลกระทบอย่างมากต่อการสังเคราะห์ทางสรีรวิทยาของฮีโมโกลบิน และโดยการทำลายเม็ดเลือดแดง จะนำไปสู่ภาวะโลหิตจาง
การจำแนกโรคธาลัสซีเมียต้องอาศัยปัจจัยสำคัญ 2 ประการคือ
- จำนวนยีนกลายพันธุ์ที่สืบทอดมาจากพ่อแม่
- ประเภทของโปรตีนที่เกี่ยวข้อง (อัลฟาหรือเบตาเฮโมโกลบิน)
อัลฟ่าธาลัสซีเมีย
ในรูปแบบ "อัลฟา" ของธาลัสซีเมีย - ซึ่งหน่วยย่อยทรงกลม 4 "อัลฟา" ของเฮโมโกลบิน (ที่โครโมโซม 16) สามารถกลายพันธุ์ได้ - ยีนที่มีข้อบกพร่องอย่างน้อยหนึ่งยีนเกี่ยวข้อง แต่ละหน่วยย่อยทรงกลมจะถูกเข้ารหัสอย่างชัดเจน จากยีน ดังนั้น ยีนที่เกี่ยวข้องได้ 4 ตัว

ภาพอาการทั่วไปจะรุนแรงขึ้นเมื่อมียีนสามหรือสี่ยีนเกี่ยวข้อง: ในกรณีแรก เราพูดถึง "โรคฮีโมโกลบิน เอช"(มีอาการปานกลางหรือรุนแรง). เมื่อยีนทั้งสี่เกี่ยวข้องกัน โรคนี้เรียกว่า อัลฟ่า-ธาลัสซีเมียเมเจอร์: ในสถานการณ์ที่คล้ายคลึงกัน เด็กแรกเกิดเสียชีวิตก่อนคลอดหรือหลังจากนั้นไม่นาน
เบต้าธาลัสซีเมีย
รูปแบบเบต้าของธาลัสซีเมียจะเกิดขึ้นเมื่อมีการกลายพันธุ์ของยีนที่เกี่ยวข้องกับองค์ประกอบของสายโซ่เบต้า (ที่ระดับโครโมโซม 11): ในกรณีนี้ ยีนเพียงสองตัวเท่านั้นที่จะได้รับผลกระทบ หากมีการเปลี่ยนแปลงยีนเพียงตัวเดียวเรียกว่า เบต้าธาลัสซีเมียไมเนอร์ซึ่งผู้ป่วยบ่นว่าไม่มีอาการที่เกี่ยวข้อง ในทำนองเดียวกันกับตัวแปรอัลฟ่า การมีส่วนร่วมของยีนทั้งสองที่ประกอบเป็นสายเบตาของเฮโมโกลบินส่งผลให้เกิดหนึ่ง เบต้าธาลัสซีเมียเมเจอร์ (หรือ โรคโลหิตจางของ Cooley) ซึ่งสะท้อนถึงอาการรุนแรงและรุนแรง อย่างไรก็ตาม ในกรณีนี้ อาการมักจะเริ่มหลังจากผ่านไปสองสามปีตั้งแต่แรกเกิด
ดูวิดีโอ
- รับชมวิดีโอบน youtube
อาการ
ข้อมูลเพิ่มเติม : อาการธาลัสซีเมีย
ธาลัสซีเมียเป็นโรคทางพันธุกรรมที่ร้ายแรงมากจนโรคธาลัสซีเมียบางชนิด เช่น อัลฟ่าธาลัสซีเมียเมเจอร์อาจทำให้ทารกเสียชีวิตระหว่างคลอดหรือหลังคลอดได้ไม่นาน อย่างไรก็ตาม ทารกที่เป็นธาลัสซีเมียเมเจอร์สามารถอยู่รอดและพัฒนาได้ อาการแรกเกิดภายในสองปีแรกเกิด (โรคโลหิตจางรุนแรง)
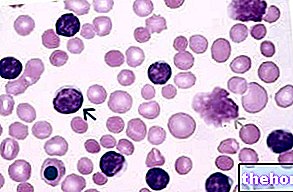
หากมีการเปลี่ยนแปลงยีนเพียงตัวเดียว ทั้งในกลุ่มอัลฟาและบีตาของธาลัสซีเมีย ผู้ป่วยจะไม่บ่นถึงอาการที่เห็นได้ชัดเจน ผ่านการวิเคราะห์ด้วยกล้องจุลทรรศน์ของตัวอย่างเลือดที่นำมาจากผู้ป่วยเท่านั้นที่มีความผิดปกติในรูปร่างและโครงสร้างของเม็ดเลือดแดงซึ่งมีขนาดเล็กกว่าปกติมาก
นอกจากโรคโลหิตจางแล้ว ผู้ป่วยธาลัสซีเมียอาจพบอาการอย่างน้อยหนึ่งอย่างต่อไปนี้: เหนื่อยล้า อารมณ์แปรปรวน (หงุดหงิด) การเจริญเติบโตล้มเหลว กระดูกใบหน้าผิดรูป ดีซ่าน หายใจลำบาก และปัสสาวะสีเข้ม
ในกรณีที่มีความรุนแรง ภาพแสดงอาการของผู้ป่วยที่เป็นโรคธาลัสซีเมียอาจเสื่อมโทรม จนถึงจุดที่ทำให้กระดูกผิดรูปโดยเฉพาะบริเวณใบหน้าและกะโหลกศีรษะ ธาลัสซีเมียสามารถส่งเสริม "การขยายตัวของไขกระดูกอย่างผิดปกติ ทั้งโดยการทำให้มวลกระดูกเปราะบางและโดยการเพิ่มความเสี่ยงของกระดูกหักอย่างมหาศาล
ท่ามกลางภาวะแทรกซ้อนของธาลัสซีเมีย ควรกล่าวถึงการสะสมของธาตุเหล็ก (ฮีโมโครมาโตซิส) ที่เป็นไปได้ การแสดงออกทั้งของโรคเองและการถ่ายเลือดซ้ำที่ผู้ป่วยต้องการ
ธาลัสซีเมียมักทำให้เกิดม้ามโต กล่าวคือ การเพิ่มปริมาตรในม้ามเกินจริง: บ่อยครั้ง ภาวะทางคลินิกทางพยาธิวิทยานี้จำเป็นต้องตัดม้าม การผ่าตัดเอาอวัยวะออก ดังที่เราทราบ ม้ามเป็นอวัยวะสำคัญที่ใช้สำหรับการสังเคราะห์เซลล์เม็ดเลือด และแอนติบอดี นอกเหนือจากการควบคุมการติดเชื้อ: การกำจัด เห็นได้ชัดว่าช่วยลดฟังก์ชันการป้องกันการดูหมิ่นของแบคทีเรียและไวรัสทำให้ผู้รับการทดลองมีความไวต่อการติดเชื้อมากขึ้น อย่างไรก็ตาม ควรชี้ให้เห็นว่าธาลัสซีเมียเองก็เพิ่มความเสี่ยงของ การติดเชื้อ: ในกรณีที่ม้ามตัดตอนในบริบทของธาลัสซีเมีย โอกาสของการติดเชื้อจะเพิ่มขึ้นเกินจริง

การวินิจฉัย
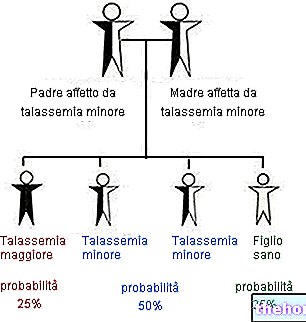
หากบิดาและ/หรือมารดาได้รับผลกระทบจากธาลัสซีเมีย โอกาสแพร่โรคสู่ลูกหลานมีสูงมากเราได้วิเคราะห์ว่าไม่ใช่ทุกรูปแบบของธาลัสซีเมียที่เริ่มต้นด้วยอาการที่ถูกต้องตั้งแต่แรกเกิด: ในสถานการณ์ที่คล้ายคลึงกัน ในกรณีที่สงสัยว่าเป็นธาลัสซีเมีย ผู้ป่วยอาจได้รับการทดสอบและการตรวจเฉพาะชุดโดยมุ่งเป้าไปที่การประเมินการวินิจฉัย ( เช่น การกำหนดเฮโมโกลบิน A2 ซึ่งเพิ่มสูงขึ้นในอาสาสมัครที่มีสุขภาพดีซึ่งมียีนเบต้าธาลัสซีมิก)
ในระหว่างการตรวจร่างกาย การคลำของม้ามในบางครั้งสามารถตรวจพบธาลัสซีเมียได้: ม้ามโตตามที่กล่าวไว้ก่อนหน้านี้ถือเป็นสัญญาณเตือนครั้งแรกสำหรับโรคโลหิตจางในทะเลเมดิเตอร์เรเนียน การตรวจเลือดมีความเฉพาะเจาะจงและแม่นยำยิ่งขึ้น: ในตัวอย่างเลือดจากธาลัสซีเมีย เซลล์เม็ดเลือดแดงเมื่อมองด้วยกล้องจุลทรรศน์จะดูเล็กและมีรูปร่างผิดปกติ นอกจากนี้ การนับเม็ดเลือดอย่างระมัดระวังของผู้ป่วยที่เป็นโรคธาลัสซีเมียจะเผยให้เห็นภาวะโลหิตจางอย่างรุนแรง: การทดสอบนี้มีประโยชน์สำหรับการนับธาตุเหล็กในเลือด เพื่อทำการวิเคราะห์ดีเอ็นเอสำหรับการประเมินการวินิจฉัยโรค และเพื่อประเมินการกลายพันธุ์ที่เป็นไปได้ของ "ฮีโมโกลบิน" .
ในทางกลับกัน อิเล็กโตรโฟรีซิสของเฮโมโกลบินเผยให้เห็นรูปร่างที่ผิดปกติของโปรตีนที่นำพาออกซิเจน
โรคธาลัสซีเมียบางชนิดไม่สามารถวินิจฉัยด้วยอิเล็กโตรโฟรีซิสได้ ในกรณีนี้ ผู้ป่วยจะต้องได้รับการทดสอบ "การวิเคราะห์การกลายพันธุ์" ซึ่งมีประโยชน์ในการตรวจหาและตรวจหาธาลัสซีเมีย
ยาและการรักษา
ดูเพิ่มเติม: ยารักษาโรคธาลัสซีเมีย
การเป็นโรคติดต่อทางพันธุกรรม เป็นที่เข้าใจได้ว่า - ในขณะนี้ - ไม่มียาใดที่สามารถย้อนกลับโรคได้ อย่างไรก็ตาม สามารถควบคุมอาการได้ ทำให้คุณภาพชีวิตของผู้ป่วยดีขึ้น การเลือกวิธีการรักษาแบบใดแบบหนึ่งมากกว่าวิธีอื่นขึ้นอยู่กับชนิดของธาลัสซีเมียและความรุนแรงของอาการ
ในโรคธาลัสซีเมียที่ไม่รุนแรง (เช่น ยีนเพียงตัวเดียวที่เปลี่ยนแปลง) ไม่จำเป็นต้องใช้ยา เนื่องจากผู้ป่วยไม่บ่นถึงอาการ ในกรณีเช่นนี้ ขอแนะนำให้ดำเนินการตรวจสอบที่จำเป็นอย่างสม่ำเสมอ บางครั้งการถ่ายเลือดเป็นครั้งคราวก็มีประโยชน์ (โดยเฉพาะในกรณีของการผ่าตัดและการคลอดบุตร)
สำหรับรูปแบบอาการในระดับปานกลางหรือรุนแรง วิธีการรักษาจะแตกต่างออกไป และอาจจำเป็นต้องได้รับการถ่ายเลือดบ่อยครั้ง หรือในกรณีที่รุนแรง จะต้องปลูกถ่ายสเต็มเซลล์
- การถ่ายเลือด: วิธีการรักษานี้ยังสามารถก่อให้เกิดโรคแทรกซ้อนที่ร้ายแรงได้ เนื่องจากการถ่ายเลือดบ่อยครั้งสามารถทำให้เกิดการสะสมของธาตุเหล็กในเลือด (ฮีโมโครมาโตซิส) ทางพยาธิวิทยา ซึ่งต้องการการรักษาเฉพาะที่มุ่งกำจัดการสะสมของธาตุเหล็ก หรือที่เรียกว่า คีเลเตอร์บำบัด (ร่วมกับยา เช่น ดีเฟราซิรอกซ์ และ ดีเฟอริโพรน) สำหรับข้อมูลเพิ่มเติม: อ่านบทความเกี่ยวกับยาสำหรับรักษา hemochromatosis
- การปลูกถ่ายไขกระดูก: สงวนไว้สำหรับกรณีที่ร้ายแรงที่สุด ซึ่งธาลัสซีเมียทำให้เกิดความผิดปกติอย่างรุนแรงในร่างกาย